RNA测序分析(五)从标准化到差异表达:DESeq2 实操指南

在上一期中,我们使用 featureCounts 获得了每个基因在各个样本中的原始 read 计数表。但这些 原始计数(raw counts) 不能直接用于比较不同样本间的表达差异——因为不同样本的测序深度、RNA 提取量、文库构建效率等因素都会造成偏差。
因此,标准化(Normalization) 与 差异表达分析(Differential Expression Analysis) 是 RNA-seq 分析中至关重要的两步。而在众多分析工具中,DESeq2 可以说是最经典、最稳定、也最常用的一款。
一、为什么需要标准化?
RNA-seq 测得的 count 并不只反映基因真实表达量,还受到技术因素影响。比如:
样本 A 测得 4000 万 reads,而样本 B 测得 6000 万 reads
如果不做标准化,B 的所有基因 count 都可能偏高
DESeq2 通过估计size factor 来校正这种差异,使得样本间的表达量可比。
二、DESeq2 的分析逻辑
DESeq2 的核心流程非常清晰:
输入原始 counts 矩阵(推荐用 featureCounts 的输出结果)
三、DESeq2 实操代码
以下为最基础的单因素差异分析流程:
1、准备数据:表达矩阵 + 样本信息
差异分析的第一步,是准备好 基因表达矩阵(featureCounts 输出的计数表)和 样本信息(metadata,包括分组信息等)。
# 读取 featureCounts 结果countData <- read.table("gene_counts.txt", header = TRUE, row.names = 1, comment.char = "#") countData <- countData[, 6:ncol(countData)] # 去掉注释列# 样本分组信息 condition <- factor(c(rep("Group1", 3), rep("Group2", 3))) colData <- data.frame(row.names = colnames(countData), condition)
metadata 中的分组信息非常重要,分析结果的准确性直接依赖于它。
2、创建 DESeq2 对象并分析差异
DESeq2 的核心流程包括 标准化 → 建模 → 差异检验:
library(DESeq2)# 创建 DESeq2 对象dds <- DESeqDataSetFromMatrix(countData = countData, colData = colData, design = ~ condition) # 标准化与差异分析 dds <- DESeq(dds) # 提取结果并按校正 P 值排序 res <- results(dds) res <- res[order(res$padj), ] write.csv(as.data.frame(res), file = "DESeq2_results.csv") # 查看显著差异基因数量 summary(res)
🔹 小提示:通常用 padj < 0.05 判断显著性,log2FoldChange 可判断基因上调或下调。
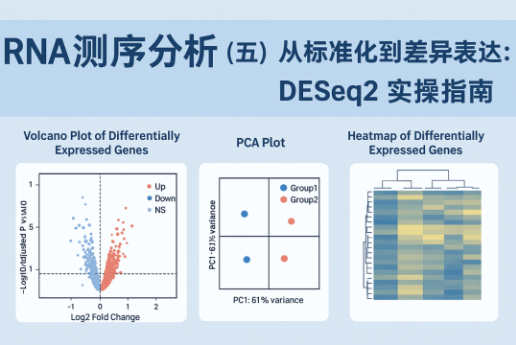
3、PCA 图:检查样本整体差异
主成分分析(PCA)可以帮助了解样本之间的整体差异,看看分组是否明显分开:
library(ggplot2)# VST 转换 vsd <- vst(dds, blind = FALSE) # 提取 PCA 数据 pcaData <- plotPCA(vsd, intgroup="condition", returnData=TRUE) percentVar <- round(100 * attr(pcaData, "percentVar")) # 绘制 PCA 图 ggplot(pcaData, aes(PC1, PC2, color=condition, shape=condition)) + geom_point(size=4) + xlab(paste0("PC1: ", percentVar[1], "% variance")) + ylab(paste0("PC2: ", percentVar[2], "% variance")) + theme_bw() + theme(text=element_text(size=14), legend.title=element_blank(), legend.position="right")
🔹 如果样本按组别明显聚集,说明分组差异明显,数据质量较好。
4、火山图:直观展示差异基因
为了让结果更直观,可以绘制 火山图(Volcano Plot):
library(ggplot2)library(dplyr)res$threshold <- as.factor(ifelse( res$padj < 0.05 & abs(res$log2FoldChange) >= 1, ifelse(res$log2FoldChange > 1, "Up", "Down"), "NS" )) res$gene <- rownames(res) ggplot(res, aes(x = log2FoldChange, y = -log10(padj), color = threshold)) + geom_point(alpha = 0.9, size = 1.2) + scale_color_manual(values = c("Up"="#D32F2F", "Down"="#1976D2", "NS"="grey70")) + geom_vline(xintercept = c(-1, 1), linetype="dashed", color="grey50") + geom_hline(yintercept = -log10(0.05), linetype="dashed", color="grey50") + theme_classic(base_size = 14) + labs(x="Log2 Fold Change", y="-Log10 Adjusted P-value", title="Volcano Plot of Differentially Expressed Genes")
🔹 红色点 = 上调基因,蓝色点 = 下调基因,灰色 = 无显著变化。火山图能快速告诉你,哪些基因在两个条件间变化最大。
差异基因热图聚类
热图可以展示显著差异基因在样本间的表达模式,并通过聚类观察相似样本和基因的分组关系。
library(pheatmap) # 筛选显著差异基因 sigGenes <- rownames(res)[which(res$padj < 0.05 & !is.na(res$padj))] # VST 转换后的表达矩阵 vsd_mat <- assay(vsd)[sigGenes, ] # 对每个基因做 Z-score 标准化 vsd_z <- t(scale(t(vsd_mat))) # 蓝黄莫兰迪配色(柔和渐变) morandi_blueyellow <- colorRampPalette(c("#A8C0D9", "#E6E2C3", "#FFDDA3"))(100) # 绘制热图 pheatmap(vsd_z, color = morandi_blueyellow, cluster_rows = TRUE, cluster_cols = TRUE, show_rownames = FALSE, show_colnames = TRUE, fontsize = 12, border_color = NA, main = "Heatmap of Differentially Expressed Genes")
五、总结
通过 DESeq2,我们可以轻松实现:
DESeq2 是 RNA-seq 分析中不可或缺的利器,让我们从海量数据中,快速锁定有生物学意义的基因。